
Trashing the safety data
Also available on Substack – Come and join the conversation with open comments
Randomised controlled trials are carried out to give an unbiased measure of the benefits and risks of a drug. To ensure that those running the trial and the participants are not biased by knowing whether or not they were treated, these trials are best performed while keeping all parties in the dark about who had treatment or placebo. If the participants are unaware (but others – eg the supervising doctors (known as “investigators”)- know which treatment has been given) it is called single blinding and if the investigators are unaware then it is referred to as double blinding.
The Pfizer/BioNTech trial claimed to be “observer blinded” only, meaning that those analysing the data were unaware who had had the treatment. The protocol said “The study staff receiving, storing, dispensing, preparing, and administering the study interventions were unblinded” and went on to say that the study manager, clinical research associate, medical monitors, clinicians, statisticians and a submission team would all be unblinded too.
The risk of having an unblinded study comes not only from what the participants report back to the people running the trial in terms of their health, but also the threshold of the people running the trial for recording such an event, as well as the detail of such recording, for example in terms of whether they attribute adverse events to the drug as opposed to being coincidental. Would covid-like symptoms be dismissed as vaccine adverse reactions in the treatment group but carefully recorded with extensive testing in the placebo group?
The people in the trial had a really important role to play. As well as demonstrating any vaccine benefit this was the best chance of getting a measurement for any harm. However, in order to measure harm it was imperative that the placebo group remain untreated for a long period. When Pandemrix vaccine caused narcolepsy in teenagers in 2009 and 2010, it took on average 8 months from injection to diagnosis. Therefore, a few months could never be an adequate period of follow up.
The FDA (in a rare display of diligence) have specified that they do “not consider availability of a COVID-19 vaccine under EUA [Emergency Use Authorisation], in and of itself, as grounds for stopping blinded follow-up in an ongoing clinical trial.” They went on, “An EUA request should include strategies that will be implemented to ensure that ongoing clinical trials of the vaccine are able to assess long term safety and efficacy.”
There was a concern that as participants became eligible for vaccination, they would drop out of the study in order to try and make sure they’d had what they believed was the active product that would protect them (rather than only placebo). Arguments about the ethical justification for crossover were focused solely on efficacy data with no mention of safety data. Fauci and others proposed a blinded crossover where each group would be reinjected while remaining unaware of the order in which they had received placebo or product. Professor Steven Goodman, Assoc. Dean, Clinical and Translational Research and Professor of Epidemiology & Population Health and Medicine at Stanford University, presented on the ethics to the FDA on 10th December (figure 1). Although such a crossover would effectively end the period over which safety data between active and placebo groups could be compared, there might still be a possibility of measuring efficacy, and the time until any waning occurred. This proposal was dismissed by the pharmaceutical companies as “onerous”.

Figure 1: Slide from Steven Goodman’s presentation to FDA
On 10th December 2020 the FDA issued an emergency use authorization for the Pfizer / BioNTech product. They said “FDA does not consider availability of a COVID-19 vaccine under EUA, in and of itself, as grounds for immediately stopping blinded follow-up in an ongoing clinical trial or grounds for offering vaccine to all placebo recipients.”
But it was too late.
Pfizer had said in October 2020, that it “would have an ethical responsibility to inform all study participants about the availability of an emergency authorized vaccine.” Pfizer had written to the participants in their trial on 11th November indicating their intention of unblinding them and offering vaccine to the placebo group (figure 2).

Figure 2: Letter to trial participants in Pfizer/BioNTech trial
They amended their protocol to allow for injecting the placebo group (with the active product) on 1st December 2020. On 14th December the first placebo recipients were injected with the product. On 12th January, the US president extended eligibility to those aged over 65 years. By then 20% of the 16-55 year olds in the trial had already been injected (figure 3). By 13th March 2021, 89% of the placebo group had been unblinded and injected and 85% had had two doses.
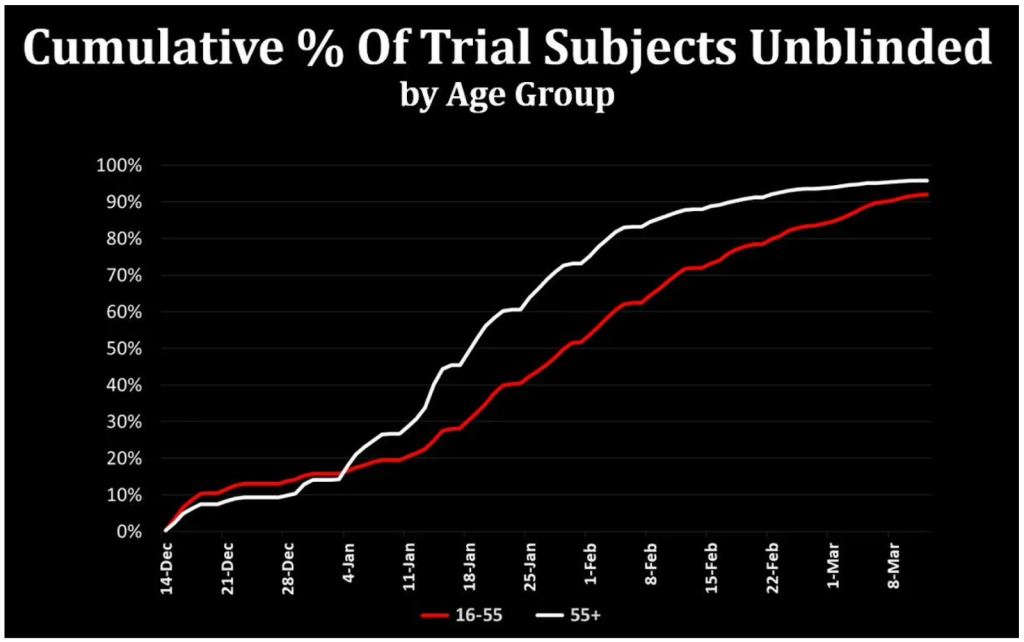
Figure 3 Percentage of participants who had been injected over time (thanks to Josh Guetzkow).
For safety follow-up a placebo group is needed to demonstrate the level of coincidental health problems. Using that baseline any additional problems caused by the product can be compared. Participants in the Pfizer/BioNTech trial had safety follow-up after the second dose of between only 3 and 140 days prior to being injected with the product. The average was only 97 days. That is all the safety data in comparison to a control group that will ever be available for these novel products, and is way below the 12 months minimum recommended by the International Coalition of Medicines Regulatory Authorities.
The argument that participants would be tempted to drop out of the trial in order to be vaccinated might ultimately have been valid, however, the participants were injected long before that risk could materialise. At the point where 89% had been injected, 37% of the US population had been injected as part of the wider rollout with much lower figures for the other sites (figure 4 and table 1).
| Percentage of trial participants at that site | Doses given per 100 people by 13th March 2021 | |
| USA | 76.2 | 36.9 |
| Brazil | 6.6 | 12.8 |
| Argentina | 13.1 | 11.4 |
| Turkey | 1.1 | 6.1 |
| Germany | 1.1 | 5.6 |
| South Africa | 1.8 | 0.2 |
Table 1: Data on proportion of participants by site and doses given in those countries by cutoff
The rollout prioritised the old and frail but younger age groups could have been eligible earlier if they were sick or were healthcare workers. The trial included “Adults 16 years of age or older who were healthy or had stable chronic medical conditions, including but not limited to human immunodeficiency virus (HIV), hepatitis B virus, or hepatitis C virus infection, were eligible for participation in the trial.” Therefore they were only likely to be prioritised if they were healthcare workers.
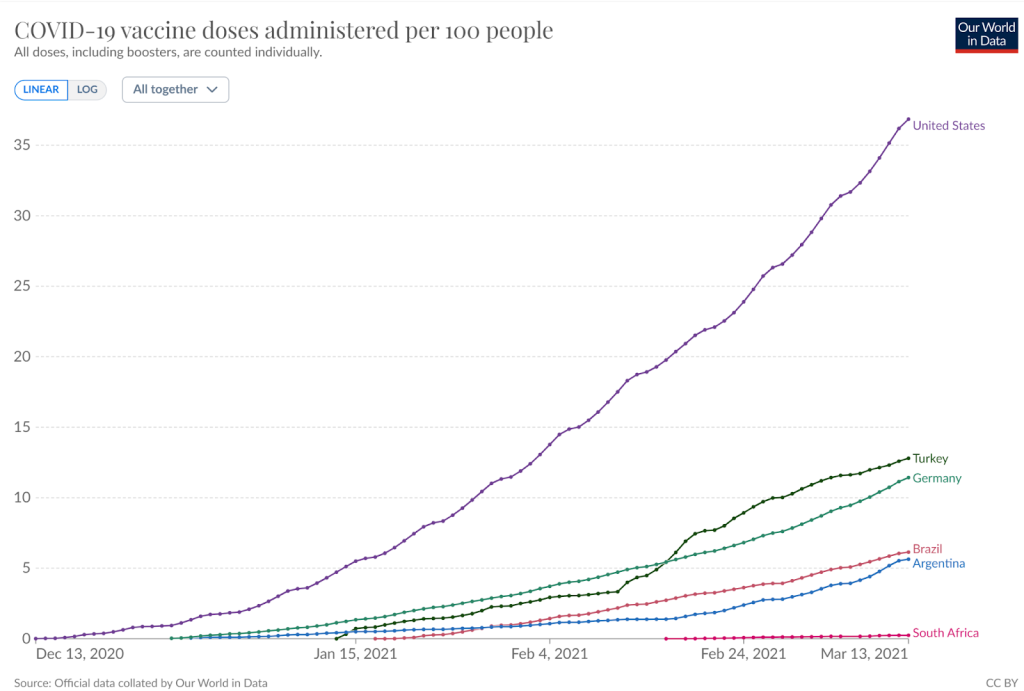
Figure 4 Percentage of population injected in each of trial site locations
For safety follow-up a placebo group is needed to demonstrate the level of coincidental health problems. Using that baseline any additional problems caused by the product can be compared. Participants in the Pfizer/BioNTech trial had safety follow-up after the second dose of between only 3 and 140 days prior to being injected with the product. The average was only 97 days. That is all the safety data in comparison to a control group that will ever be available for these novel products, and is way below the 12 months minimum recommended by the International Coalition of Medicines Regulatory Authorities.
Moderna had also contacted participants offering them a chance to unblind themselves and be injected with the product if they were placebo recipients. This offer was made on 17th December before the FDA had given any authorisation of their product.
The pharmaceutical companies were clearly eager to inject the placebo group. To justify this, they concocted a variety of “ethical” reasons of dubious credibility, leaving the suspicion that the real reason for this was to deliberately lose the ability to do a properly-controlled quantitative safety analysis. The speed with which this was carried out was unprecedented and totally unjustified, and the regulators utterly failed to hold the pharmaceutical companies to account for failing to conduct their trial in a manner which allowed it to answer key questions about their products.
