

A triumph of hope over evidence
The MHRA have recently approved novel “vaccines” that claim to protect against Omicron BA4/BA5 variants. The evidence used for the basis of these approvals is paper thin and the lack of transparency has worsened recently.
There are now three “vaccines” using different sequences of RNA. The first were those using the Wuhan sequence. People continued to be injected with these for 21 months after the Wuhan wave peaked. Manufacturers then developed “vaccines” containing sequences of Omicron BA1. For unfathomable reasons, these newer products also contained the Wuhan sequence. The latest products contain sequences to BA4/BA5 variants.
How much testing was done on these products?
The Wuhan products were tested in large clinical trials. There were several serious issues with these trials but, taken at face value, they demonstrated a reduction in the number of symptomatic people who tested positive for covid. However, since then, properly controlled trials of vaccine efficacy “in the field” have not been carried out.
The BA1 products were approved on the basis of the levels of antibody produced. No-one has ever demonstrated a level of antibody in the blood which provides protection against covid infection. The government wording says, the product aims “to generate neutralising antibodies, which may contribute to protection against COVID-19.” This is not based on any scientific evidence, merely hope.
Moderna BA1
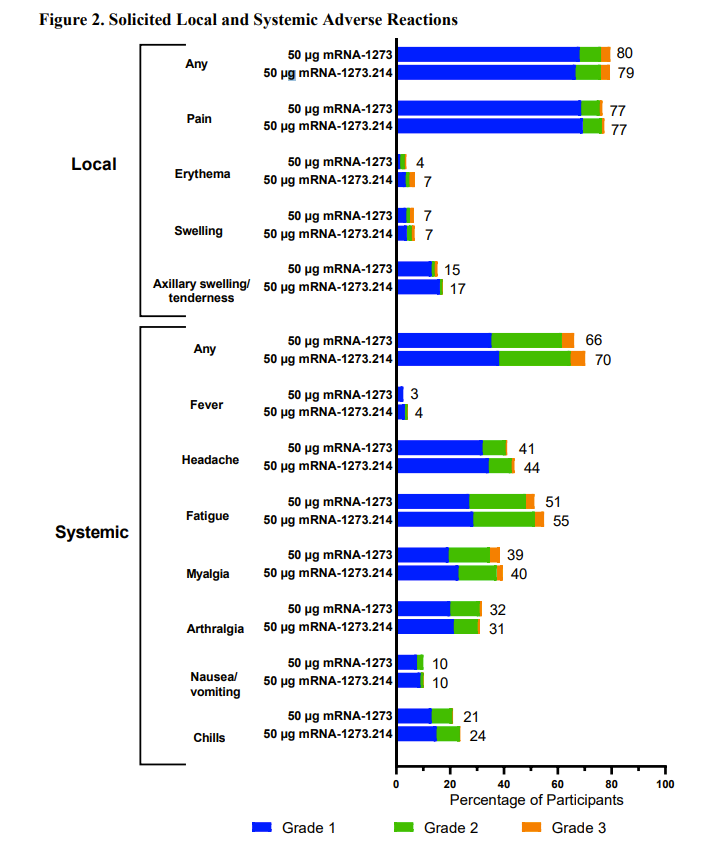
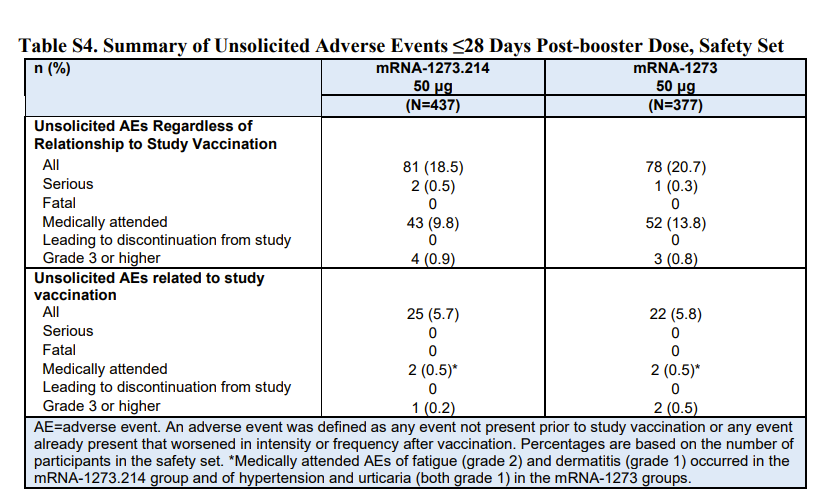
For Moderna, the BA1 (accompanied by Wuhan) product was tested in 437 people who were compared to 377 people given a fourth dose of the original product. A rise in antibody levels was demonstrated in both groups and the new product was then declared a success. However, the underlying study also looked at infections. Of those given the Moderna BA1 booster there were 11 who tested positive for covid after more than 14 days from vaccination and 5 of these were symptomatic. That compared to only 5 in the original Moderna group, only one of whom was symptomatic. These findings appear to have been ignored even though they were published alongside a table that the MHRA replicated in their own analysis.
As well as there being at least twice as much covid in the Moderna BA1 group than in the original Moderna group, there was also a worse safety profile. Almost every systemic side effect was reported more often for the BA1 booster. The MHRA decided that because the safety signals were similar to a booster of original Moderna and similar to the first dose of Moderna they could conclude, “No new safety signals were identified.” One in ten people who received the Moderna BA1 product had to consult with a doctor within a month. That was indeed a similar number to the original product, but it is still concerningly high. Nevertheless the MHRA authorised the product “under a so-called ‘conditional approval’ scheme.”
Pfizer-BioNTech BA1
The Pfizer-BioNTech BA1 product was tested on 610 people who had had three doses of the Wuhan product. They were divided into a group that received a fourth Wuhan product and a group that received the Pfizer-BioNTech BA1 product (305 in each group). The results for almost half of these people are not disclosed, nor are the reasons why the subjects were excluded from either group given, which would ordinarily be regarded as essential in evaluating the results. The table showing the rise in antibodies (table 2) shows the results for only between 50% and 60% of the people given the product. It is worth noting that the levels of antibody to the original strain are almost ten times higher than the levels produced against Omicron after the Pfizer-BioNTech BA1 product. No data from the trials is available to the public.
The MHRA produces safety Public Assessment reports which are said to “explain our assessment of the scientific evidence used to lead to regulatory decisions on the safety of medicines or medicines classes.” The release of the Public Assessment report for the Pfizer-BioNTech BA1 product contained no research evidence at all. It said, “MHRA decided that the benefits are greater than the risks and recommended that this medicine/these medicines can be approved for use.” and “The quality of the product is acceptable, and no new non-clinical or clinical safety concerns have been identified. The benefit/risk balance is, therefore, considered to be positive.”
Again safety was only measured for conditions the manufacturer included in their survey. It was compared to previous doses and then waved through because there were “no new adverse reactions.”

The latest products
As if the above was not bad enough this is where things get truly ridiculous. The Omicron BA.1 variant has been obsolete since early 2022 and the manufacturers have since produced products for more recent (also now obsolete) Omicron variants BA4 and BA5.
Moderna BA.4/BA.5
The only data on this product was from testing 10 mice and measuring their antibody levels and showing they were similar to mice given BA1. All 10 contracted covid after receiving the product. The MHRA have not yet approved this product but the FDA have, and they based their decision entirely on what they hope might happen, extrapolating from what prior products were thought to achieve.
Pfizer-BioNTech BA.4/BA.5
The only data on this product was obtained from studying 8 mice and measuring their antibody levels, and showing they were similar to mice given BA.1.
There has been no human trial carried out by the manufacturers. The decision to approve was based entirely on extrapolating from the data used to approve previous vaccines (data which is not in the public domain for the Pfizer-BioNTech BA.1 product). Again, this is using hope, not science, to support decision making.
A study the MHRA did not reference has shown that a quarter of healthcare workers could not attend work after receiving the Pfizer-BioNTech BA.4/BA.5 product compared to 10% for the original product. 85% had an adverse reaction of some kind compared to 51% for the original product.
The MHRA said, “The updated booster vaccine is the second bivalent vaccine from Pfizer/BioNTech to receive MHRA approval, after it was found to meet the UK regulator’s standards of safety, quality and effectiveness. The vaccine has been approved for use as a booster dose in individuals aged 12 years and above…The MHRA’s decision is based on all available evidence on the original Pfizer/BioNTech COVID-19 vaccine and its adapted vaccines”
The MHRA closes their statement by declaring this easily refuted belief: “A recent paper in The Lancet journal suggested that COVID-19 vaccines have prevented up to 20 million deaths in the first year of use.” This particular hope is at the level of a fantasy.
The idea behind using sequences from Omicron in these latest products is to ensure that people have immunity to the most recent circulating variant. However, for both Pfizer-BioNTech and Moderna the MHRA have said that these products should not be given to people who have not had a primary course of the outdated Wuhan sequence. Why not?
Two years after first approvals were given the MHRA have still not requested genotoxicity or carcinogenicity studies, merely stating that “The components of the vaccine (lipids and mRNA) are not expected to have genotoxic potential.” Another hope that, unfortunately, may ultimately prove not be substantiated.
As the evidence has diminished with each iteration so has the transparency
Public Assessment Reports are published by the MHRA to share the data and rationale that supported their decision making. Public Assessment Reports were published 6 days after approval for the Astra Zeneca vaccine, 13 days later for the original Pfizer-BioNTech vaccine & 6 weeks later for the original Moderna. Moderna was approved in August 2022 and Pfizer-BioNTech in September 2022. There was a 13 and 8 week gap respectively for the latest reports to be published.
On 16th September 2022, an FOI request was made for these reports. The MHRA replied saying they were exempt from sharing as they were due to be published publicly in 8 weeks. Those 8 weeks have passed with no publication. They said in their response, “We recognise there is strong interest in seeing this data and accept it should not be withheld. We believe that the public interest will be best served by accompanying the publication of the data with an analysis to aid its interpretation which is currently under preparation.” Why do the public need such an analysis? None were provided for the reports on earlier vaccines.
Thanks to an intervention by the information commission who gave the MHRA until 1st December to respond, these have now been published.
Other FOIs have been raised to request the mRNA sequence for the bivalent boosters. The MHRA supplied the Pfizer sequence in full. However, the Moderna sequence was completely redacted “due to commercial interests.” Why is there a lack of transparency and why is one pharmaceutical company treated differently to another for a similar product?
Conclusion
The MHRA is 86% funded including from user fees paid by pharmaceutical companies and as that percentage has risen the proportion of applications that have been approved has risen in line. Dr June Raine the Chief Executive of the MHRA has publicly declared that she unilaterally changed the role of the regulator from “watchdog to enabler.”
The regulator has a role in ensuring medical products that do not provide net benefit are not approved, but their main aim is to ensure the safety of the public. They are in a powerful position able to demand rigorous evidence of safety. Instead of doing so, the evidence required to demonstrate efficacy has declined to reports of antibody levels (rather than lack of disease) in a handful of mice, and as regards safety, this is based on a mere assumption that evidence from other products can be extrapolated. Part of their justification for this low bar seems to be their belief in the modelled fantasy of 20 million lives having been saved by vaccination. Their willingness to believe shoddy evidence from mice data stands in sharp contrast to their unwillingness to listen to the testimony of the vaccine injured and engage with them properly.
